Challenges in quantum chemistry
The study of microscopic systems, in particular molecules, revolves around the resolution of the Schrödinger equation – a problem being tackled by many researchers in the quantum chemistry community. Solving the Schrödinger equation exactly is difficult, i.e. the difficulty increases exponentially with the number of electrons in the system. Chemists must rely on computational techniques and clever approximations to address the demands of the problem.
The history of computational chemistry is deeply intertwined with computer hardware development, where computer science and electrical engineering play a predominant role. Understanding electron-electron interactions is crucial to accurately model chemical properties and the behavior of frontier orbitals, i.e. molecular orbitals usually involved in chemical reactions. However, it is this aspect that makes quantum chemistry complicated (or hard). The dimension of the wavefunction describing the system grows exponentially with the system size, making industrially-relevant molecules arduous (if not impossible) to study via quantum mechanics and classical computing. This is where quantum computers could shine, as storing quantum information on quantum bits (qubits) is far more efficient than on classical bits.
One of the best-positioned fields to benefit from advances in quantum computing is chemistry, as detailed in a recent Chemical Reviews article. However, there is still much to be discovered on the algorithms side, as quantum computing is a nascent, rapidly-evolving technology. To this end, we have developed Tangelo to enable chemists to easily leverage the power of quantum computing for chemistry.
Enter Tangelo
Friction brings about the spark of innovation, and by enabling scientists to conveniently implement the latest algorithms and reproduce published results, they can challenge new claims in advancing research. We built Tangelo with exactly this in mind, as an open-source python package focusing on the development of material simulation workflows on quantum computers.
Tangelo’s purpose is to accelerate R&D by equipping researchers with a convenient collection of reusable building blocks and algorithms. The package provides a framework to innovate on the various steps of quantum computing workflows – for example, state preparation or error mitigation – while taking advantage of existing frameworks (Braket, Qiskit, Cirq, etc). Users are empowered to explore their own avenues by drawing components from these modular, reusable and extensible toolboxes (Fig. 1).
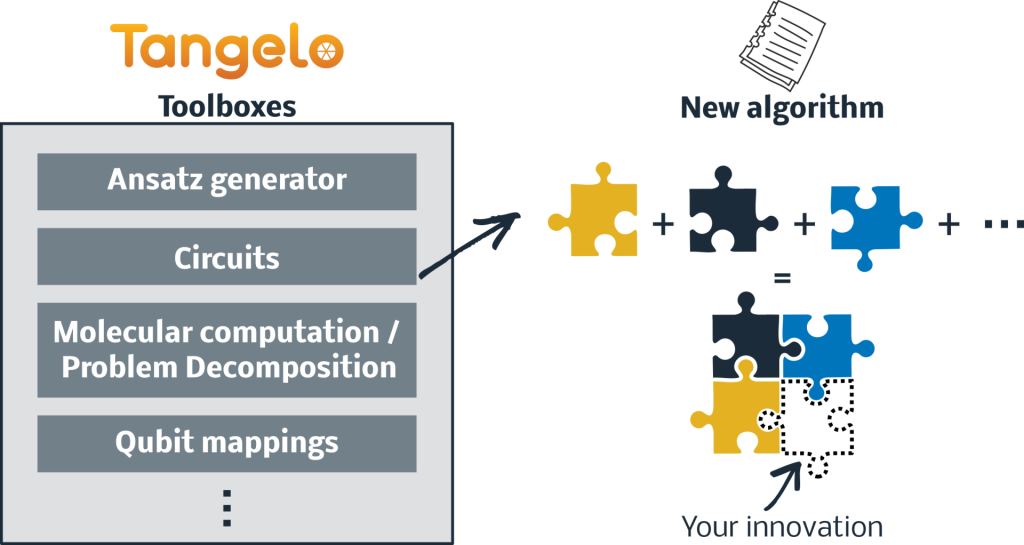
We highlight two examples below of the quantum capabilities enabled by Tangelo. The first is a Noisy Intermediate-Scale Quantum (NISQ) application, applicable to today’s devices that have limited qubits and circuit depth. The other involves a method applicable to future fault-tolerant quantum computing, which could accelerate the discovery of materials and new drugs.
Exploring NISQ algorithms with Tangelo
Noisy Intermediate-Scale Quantum (NISQ) devices are a class of quantum hardware that are currently available, but suffer from limited qubit connectivity, coherence times, and noise. Developing efficient and effective algorithms for these devices remains a challenge in solving large-scale problems.
By breaking down a large problem into smaller sub-problems, it is possible to solve larger quantum chemistry problems on NISQ devices while minimizing the impact of noise and errors. This approach requires additional steps, but is an essential step towards enabling the solution of complex quantum chemistry problems on NISQ devices.
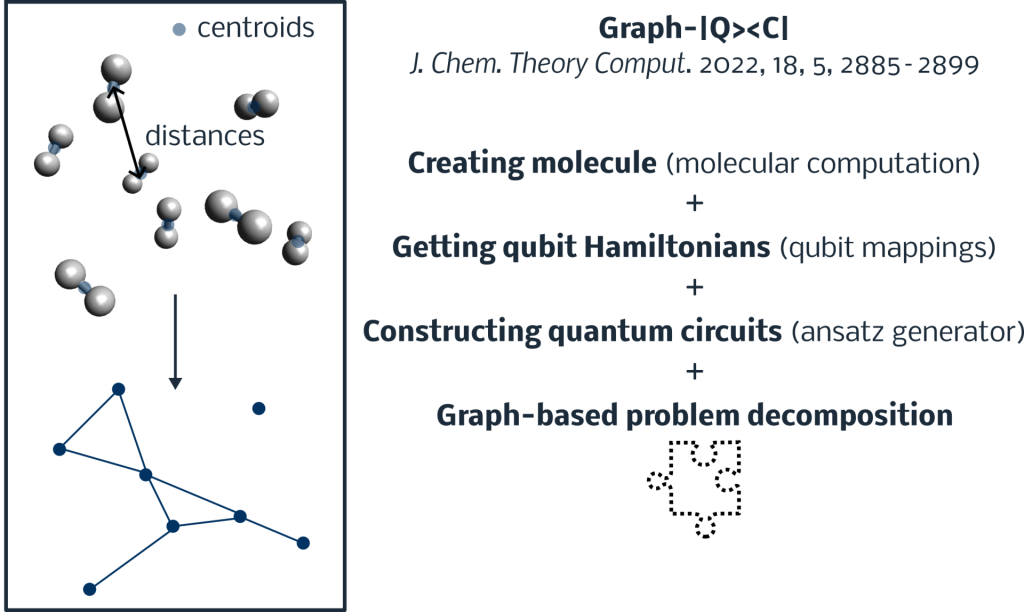
At Good Chemistry, we have a history of successfully using problem decomposition methods to scale-up quantum chemistry computation via massive parallelization. This top-down approach has also been used to optimize an electronic structure calculation on a trapped-ion quantum computer. In the same vein is the hybrid Graph-|Q><C| algorithm, published by Indiana University researchers in 2022.
Graph-|Q><C| divides a molecular system into many fragments using a graphing procedure, as shown in Figure 2. This algorithm, which can be viewed as an extension of the ONIOM method, computes the summation of energy differences from high- and low-accuracy quantum chemistry electronic structure approaches. Here, high (resp. low) accuracy methods refer to increasing (resp. decreasing) the ansatz expressibility or dealing with bigger (resp. smaller) basis sets. By reusing the ONIOM and molecular computation tools inside Tangelo, a user is in a position to reproduce and go beyond the published results with little effort. We were able to accomplish this with about 56 lines of code, excluding documentation and reference computation!
To fault-tolerant quantum computing and beyond
Fault-tolerant quantum computing is where many people expect to observe meaningful quantum advantages over classical computing. Generally, fault-tolerant methods require hardware that goes beyond the limitations of current NISQ devices.
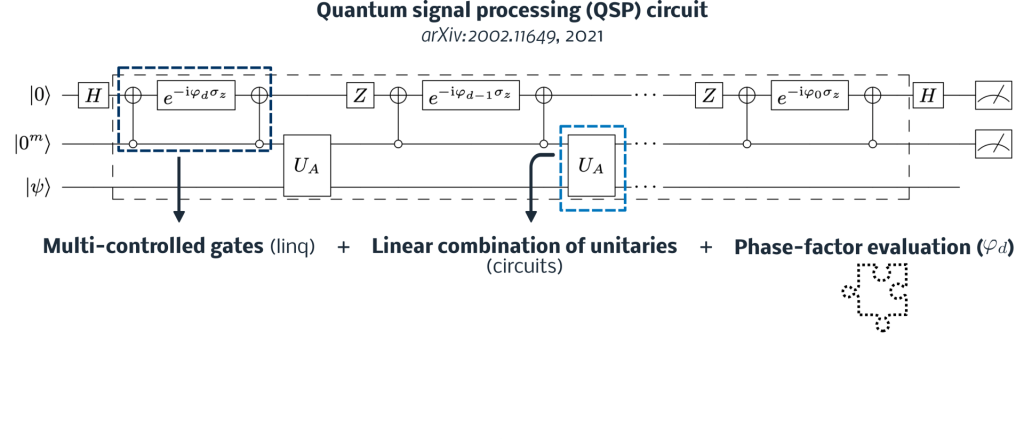
Quantum signal processing (QSP) is one of these methods, where eigenvalues of a Hamiltonian are mapped to ancilla qubits. After applying single-qubit rotations to this register, a projection-based measurement is made to apply – with a certain probability threshold – a polynomial in terms of the Hamiltonian to a quantum state (Fig. 3). This polynomial can be defined to perform Hamiltonian Simulation, eigenstate filtering, matrix inversion, and much more. The underlying component of QSP utilizes fundamental components like multi-controlled gates, linear combination of unitary (LCU), and mid-circuit measurement, which are all available building blocks in Tangelo. An example of constructing a QSP circuit for eigenstate filtering is available at this link.
Tangelo: Modular and versatile quantum computing for chemistry
Tangelo was designed to empower users to stay on top of quantum computing, a fast-moving state-of-the-art technology. This blog showcased just two examples, but we had many similar experiences with other innovations such as TETRIS-ADAPT-VQE, a collection of algorithms targeting excited states, pair UCCD ansatz, and more.
Modularity was always at the core of Tangelo, to maximize the hackability of quantum algorithms for chemistry applications. Tangelo has translation functions for quantum circuits and operators, allowing users to easily jump between Tangelo and other popular quantum computing frameworks (Qiskit, Cirq, Pennylane, Q#, etc.) and therefore leverage the innovation scattered across various projects.
We are thrilled to contribute to the community by open–sourcing our research tools, and believe they can help you advance the field: the license is very permissive. We welcome contributions in any form (discussions, issue reports, implementation/suggestion of new features…). In the meantime, we invite you to give us a star on github and give Tangelo a shot, as this helps us tremendously for the visibility of our project. 🍊

