The Problem of Crystal Structure Prediction
Organic Crystal Structure Prediction (CSP) refers to the problem of trying to predict the crystal structure that will form from a given molecule. Crystallization occurs when atoms or molecules start to form clusters, which can be triggered by precipitating in a solution or by freezing. These clusters are periodic – they are composed of a single unit (or unit cell) that is repeated in all directions. In an organic crystal, this repeating unit contains multiple copies of the same molecule. The molecules found in the unit cell can arrange themselves in many different configurations, leading to multiple stable physical states in which the crystallized form of a molecule may be found. Each stable state is known as a polymorph.
Knowing the number, and stability of polymorphs which can form from a given molecule has important implications in a number of industries. For example, the crystallized form of a molecule may affect the efficacy and safety of a drug. In the development of organic semiconductors for flexible electronic devices, different polymorphs can exhibit different opto-electronic performance. The ability to do accurate CSP in-silico has the potential to greatly accelerate work done in the laboratory. On top of the obvious economic benefit of such an approach, CSP also facilitates new materials discovery, allowing researchers to discover novel crystal structures with desired properties, which can then be synthesized in the lab.

Despite the widespread applications of CSP, it remains an extremely difficult problem to solve. Crystals exhibit high levels of symmetry, which can be classified into space groups – the mathematical description of how molecules in the unit cell relate to one another, with 230 distinct space groups existing in three dimensions. In addition, a crystal can have a different number of molecules which pack together in the unit cell. Lastly, and perhaps most importantly, it is exceedingly difficult to accurately quantify the stability of a polymorph, with energy differences between different polymorphs tending to be extremely small, rarely surpassing 2.5 kcal/mol, and requiring extremely accurate potentials modeled at quantum mechanical accuracy. Therefore, the problem in CSP is twofold – one needs to effectively explore a massive chemical space, while at the same time accurately rank these samples to identify lower energy polymorphs.
Due to the challenges of predicting stable polymorphs, the Cambridge Crystallographic Data Centre (CCDC) has periodically organized “blind test” challenges to encourage the development of computational tools for CSP. While the first blind test focused on small rigid molecules, the most recent blind test contained more difficult targets – bulky flexible molecules, co-crystals, and a former drug candidate which has 5 known polymorphs. The field has seen an explosion of interest, with the latest blind test receiving 25 submissions from over 100 researchers.
Although CSP methods have achieved remarkable accuracy, they are extremely costly to perform. In the latest blind test, hundreds of thousands of CPU hours were used to rank structures using potentials modeled at the quantum mechanical level. Specifically, Density Functional Theory (DFT) has proven to be effective for ranking crystal systems. However, evaluating the energy of a single crystal system using DFT takes hours of computation.
An efficient alternative to using DFT is to train a machine learning model to predict the DFT energy of an organic system. In the last few years, machine learning models which are able to predict the DFT energy of organic molecules within chemical accuracy have been developed. In contrast to DFT, it takes a few seconds to evaluate the energy of a crystal system using these models.
GAmuza: Good Chemistry’s Solution for Tackling CSP
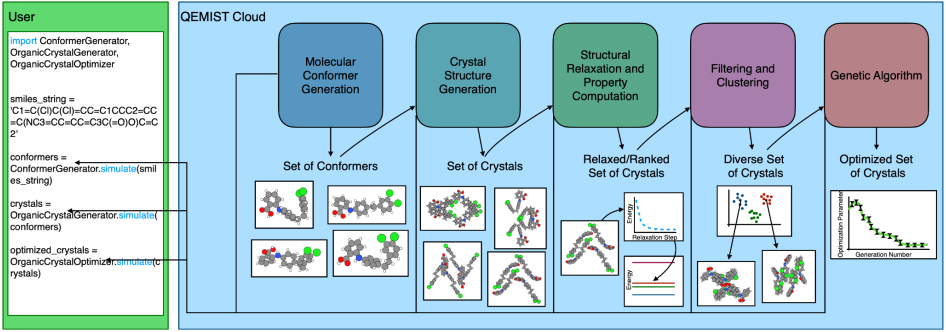
To tackle numerous challenges that arise during CSP, Good Chemistry has integrated an organic crystal structure prediction pipeline into QEMIST Cloud. Our solution solves the two major hurdles of CSP by using neural network potentials in the form of the ANI models, trained to predict the energy of molecules, to allow for fast and accurate scoring, and pairs these models with a genetic algorithm (GA) to produce diverse, high-quality samples from the vast chemical space. We call our in-house CSP tool “GAmuza”, named after a mountain peak located in the North Cascades of southwestern British Columbia.
Our pipeline begins with a random search of compatible crystal structures. Given a target molecule, our pipeline first generates the three-dimensional coordinates of the molecule in question. While rigid molecules can only be found in a single conformation, flexible molecules may have more than one molecular conformation (conformer). For each conformer, we then generate up to millions of crystal candidates. For simpler molecules, our pipeline can generate millions of candidate crystals in a matter of minutes. These candidates are then screened with a machine learning model capable of evaluating the energy of millions of crystal candidates in the span of a few hours, when run on multiple CPUs.
To more effectively sample the space of low-energy polymorphs, we use a genetic algorithm to make improvements to the population generated via random search. A genetic algorithm (GA) makes improvements to a population of candidate solutions (in our case, different crystal structures) by using biologically inspired operators to modify candidates.
The GA proceeds by adding a new structure to the population each generation. At the beginning of each generation, parents are selected to produce an offspring (i.e. a novel crystal structure) based on their fitness. In our case, fitness is measured by how low the predicted DFT energy of the candidate is according to the machine learning model. Producing new crystal structures in this manner promotes a “survival of the fittest” strategy, evolving the population towards lower energy structures.
GAmuza Achieves State-of-the-Art Results on CSP Blind Tests
To assess the capability of our tool, we tested it on 21 targets from the 6 CSP blind tests. We first showed that during random search, 10 out of the 21 targets are generated. We then put our full pipeline to the test – beginning with a population generated via random search, and evolving that population with our genetic algorithm. Machine learning models predicting the DFT energy were used to both refine the initial population created by random search, and to select parents throughout the genetic algorithm. This allowed us to generate two more targets from the blind tests. Notably, these two structures came from the two latest blind tests
Lastly, we wanted to see how our genetic algorithm performed when it has access to an idealized fitness function. In this scenario, the algorithm knows how far each structure is from the experimentally derived polymorph, and applies mutation and crossover operations on parents chosen from the population using various selection strategies to try and find a structure that is even closer to the experimental structure. This allowed us to generate another four targets from the six blind tests.
Our idealized GA performs favourably when compared to other submissions from the blind tests. GAmuza achieved a higher success rate than the average submission in all of the four latest blind tests. Furthermore, as expected, our tool achieves a significant speed advantage over tools that use DFT directly to evaluate energies. In Figure 3, We have plotted the time it takes our tool (when used with an ML potential) versus the fastest, average, and slowest tool used to generate various targets from the last three blind tests.

The results achieved by our CSP pipeline are significant. We present, for the first time, a continuously improvable scoring method – competitive in speed with the fastest methods in the literature with the potential to achieve state-of-the-art results. Our analysis also shows that there still exists a gap between machine learned potentials and DFT. Currently, the ANI models are trained on a dataset containing gas-phase molecules, and extrapolate patterns learned from this data to predict the energy of crystal systems. We hypothesize that ANI models trained on a dataset of organic crystals will drastically improve our pipeline. The future work of Good Chemistry is to improve the models to have the most efficient and accurate CSP pipeline.
Stay up to date with our work by signing up for our newsletter here, and view our corresponding manuscript on ArXiv here!


